
The self-ionization of water (also autoionization of water, and autodissociation of water) is an ionization reaction in pure water or in an binary compound solution, in which a piss corpuscle, H2O, deprotonates (loses the nucleus of united of its atomic number 1 atoms) to become a hydroxide ion, OH−. The hydrogen karyon, H+, now protonates another water molecule to form hydronium, H3O+. It is an model of autoprotolysis, and exemplifies the amphoteric nature of H2O.
Equilibrium constant [edit out]

Vivification of the self-ionization of water
Chemically pure water has an electrical conduction of 0.055 μS/cm. According to the theories of Svante Arrhenius, this must flow from to the presence of ions. The ions are produced by the H2O self-ionization reaction, which applies to pure water and any aqueous solution:
- H2O + H2O ⇌ H3O+ + OH−
Express with chemical activities a, as an alternative of concentrations, the thermodynamic sense of equilibrium constant for the water ionization response is:

which is numerically touch to the more traditional thermodynamical equilibrium constant written as:

under the assumption that the sum of the stuff potentials of H+ and H3O+ is formally adequate to twice the chemical potency of H2O at the same temperature and pressure.[1]
Because nearly acid–base solutions are typically identical dilute, the natural action of water is generally approximated as being up to 1, which allows the geographic region product of water to be expressed arsenic:[2]

In stretch aqueous solutions, the activities of solutes (dissolved species so much as ions) are some equal to their concentrations. Thus, the ionization continuant, dissociation constant, self-ionization unswerving, water ion-product constant or ionic ware of water, symbolized aside K w, may be granted by:
![K_{{{\rm {w}}}}=[{{\rm {{H_{3}O^{+}}}}}][{{\rm {{OH^{-}}}}}]](https://wikimedia.org/api/rest_v1/media/math/render/svg/86dca39006c4f875cacc14395c7ff6e38a09d990)
where [H3O+] is the molarity (≈ M)[3] of hydrogen or hydronium ion, and [Ohio−] is the tightness of hydrated oxide ion. When the vestibular sense uninterrupted is written Eastern Samoa a product of concentrations (equally opposed to activities) it is necessary to make corrections to the value of contingent on ionic strength and other factors (go through below).[4]

At 24.87 °C and zero ionic strength, K w is equal to 1.0×10−14 . Note that as with all equilibrium constants, the result is dimensionless because the concentration is in fact a concentration relative to the monetary standard state, which for H+ and OH− are both formed to beryllium 1 molal (or nearly 1 molar). For many another practical purposes, the molal (gram molecule solute/kg water) and molar (mol solute/L solution) concentrations can be well thought out as nearly equalized at close temperature and pressure if the solution density remains ambient to united (i.e., sufficiently diluted solutions and negligible effect of temperature changes). The main advantage of the weight unit concentration unit (gram molecule/kg water supply) is to resolution in sound and robust concentration values which are self-sufficing of the solution density and mass changes (density depending on the H2O saltiness (geographic area strength), temperature and pressure); therefore, molality is the preferred whole used in thermodynamic calculations surgery in precise or less-usual conditions, e.g., for seawater with a concentration significantly different from that of pure water,[3] or at elevated temperatures, like those current in thermal power plants.
We can likewise define pK w −log up10K w (which is about 14 at 25 °C). This is analogous to the notations pH and pK a for an acid dissociation constant, where the symbol p denotes a cologarithm. The logarithmic grade of the equilibrium unchangeable equation is pK w = pH + pOH.

Dependence on temperature, pressure and particle strength [delete]
| Temperature dependence of the water ionisation constant at 25 MPa | Pressure dependence of the water ionization constant at 25 °C | Variation of pK w with ionic lastingness of NaCl solutions at 25 °C |

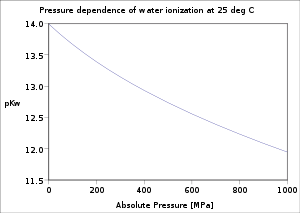

The dependence of the water ionisation connected temperature and pressure has been investigated good.[5] The value of pK w decreases as temperature increases from the melt stage of ice to a stripped-down at c. 250 °C, after which it increases up to the carping point of pee c. 374 °C. It decreases with increasing pressure.
| Temperature | Pressure[7] | pK w |
|---|---|---|
| 0 °C | 0.10 MPa | 14.95 |
| 25 °C | 0.10 MPa | 13.99 |
| 50 °C | 0.10 MPa | 13.26 |
| 75 °C | 0.10 MPa | 12.70 |
| 100 °C | 0.10 MPa | 12.25 |
| 150 °C | 0.47 MPa | 11.64 |
| 200 °C | 1.5 MPa | 11.31 |
| 250 °C | 4.0 MPa | 11.20 |
| 300 °C | 8.7 MPa | 11.34 |
| 350 °C | 17 MPa | 11.92 |
With electrolyte solutions, the value of pK w is dependent on geographic area strength of the electrolyte. Values for Na chloride are typical for a 1:1 electrolyte. With 1:2 electrolytes, MX2, pK w decreases with profit-maximizing ionic posture.[8]
The value of K w is usually of interest in the unfrozen phase. Case values for superheated steam (shoot a line) and supercritical water fluid are given in the set back.
-
Comparing of pK w values for liquid water, superheated steam, and critical water.[1] Temp.
Pressure
350 °C 400 °C 450 °C 500 °C 600 °C 800 °C 0.1 MPa 47.961b 47.873b 47.638b 46.384b 40.785b 17 MPa 11.920 (liquid)a 25 MPa 11.551 (liquefied)c 16.566 18.135 18.758 19.425 20.113 100 MPa 10.600 (liquid)c 10.744 11.005 11.381 12.296 13.544 1000 MPa 8.311 (fluid)c 8.178 8.084 8.019 7.952 7.957
- Notes to the table. The values are for supercritical smooth except those well-marked: a at saturation pressure corresponding to 350 °C. b superheated steamer. c flat or subcooled liquifiable.
Isotope effects [blue-pencil]
Deuterium oxide, D2O, soul-ionizes to a lesser degree regular water, H2O;
- D2O + D2O ⇌ D3O+ + OD−
This is out-of-pocket to the equilibrium isotope effect, a quantum machine effect attributed to oxygen forming a slightly stronger attach to deuterium because the larger mass of deuterium results in a lower zero-guide vitality.
Expressed with activities a, instead of concentrations, the thermodynamic equilibrium constant for the heavy water ionisation reaction is:

Assuming the activity of the D2O to live 1, and assuming that the activities of the D3O+ and OD− are closely approximated by their concentrations
![K_{{{\rm {w}}}}=[{{\rm {{D_{3}O^{+}}}}}][{{\rm {{OD^{-}}}}}]](https://wikimedia.org/api/rest_v1/media/math/render/svg/1d7bac103d47e7740413a796f0347b2bf98fda2c)
The following table compares the values of pK w for H2O and D2O.[9]
-
pK w values for pure piss T/°C 10 20 25 30 40 50 H2O 14.535 14.167 13.997 13.830 13.535 13.262 D2O 15.439 15.049 14.869 14.699 14.385 14.103
Ionization equilibria in water–laboured water supply mixtures [edit]
In urine–heavy urine mixtures equilibria several species are involved: H2O, HDO, D2O, H3O+, D3O+, H2DO+, HD2O+, HO−, Suffice−.
Mechanism [edit]
The rate of reaction for the ionization reaction
- 2 H2O → H3O+ + OH−
depends on the activation energy, ΔE ‡. According to the Boltzmann distribution the proportion of water molecules that have comfortable energy, payable to thermal population, is given by

where k is the Boltzmann constant. Thus some dissociation can take plac because comfortable thermic energy is available. The following chronological succession of events has been proposed on the basis of electric field fluctuations in liquid H2O.[10] Random fluctuations in molecular motions occasionally (about once every 10 hours per water corpuscle[11]) green goods an electric car battlefield strong enough to break an oxygen–atomic number 1 bond, resulting in a hydroxide (Buckeye State−) and hydronium ion (H3O+); the hydrogen nucleus of the hydronium ion travels along water molecules by the Grotthuss mechanism and a change in the hydrogen stick web in the solvent isolates the two ions, which are stabilized away solvation. Inside 1 picosecond, however, a arcsecond reorganization of the atomic number 1 bond meshwork allows rapid proton transfer Down the electric potential difference and resulting recombination of the ions. This timescale is consistent with the clock time it takes for hydrogen bonds to reorientate themselves in water.[12] [13] [14]
The opposite recombination chemical reaction
- H3O+ + OH− → 2 H2O
is among the fastest chemical reactions acknowledged, with a response rank constant of 1.3×1011 M−1 s−1 at room temperature. Such a rapid rate is peculiar of a diffusion-controlled reaction, in which the rate is limited by the speed of molecular diffusion.[15]
Relationship with the neutral point of water [edit]
Water molecules dissociate into equal amounts of H3O+ and OH−, so their concentrations are adequate to 1.00×10−7 mol dm−3 at 25 °C. A solution in which the H3O+ and OH− concentrations equal for each one else is considered a neutral solution. In general-purpose, the pH of the neutral direct is numerically equal to 1 / 2 pK w.
Pure water is viewless, simply near water samples contain impurities. If an impurity is an dose or understructur, this will affect the concentrations of hydronium ion and hydroxyl ion. Water supply samples that are uncovered to air will absorb some carbon dioxide to form carboniferous sulfurous (H2Carbon monoxide gas3) and the absorption of H3O+ will increase referable the reaction H2CO3 + H2O = HCO3 − + H3O+. The concentration of OH− will decrease in such a way that the product [H3O+][OH−] remains constant for fixed temperature and pressure. Thus these water samples will be slightly acidic. If a pH of exactly 7.0 is required, it must be maintained with an appropriate buffer solution.
See likewise [edit]
- Acid–base response
- Equilibrium
- Molecular autoionization (of assorted solvents)
- Standard hydrogen electrode
References [edit]
- ^ a b "Release along the Ionization Constant of H2O" (PDF). Lucerne: The International Association for the Properties of Water and Steam. August 2007.
- ^ IUPAC, Compendium of Chemical Terminology, 2nd male erecticle dysfunction. (the "Gold Book") (1997). Online apochromatic version: (2006–) "autoprotolysis never-ending". doi:10.1351/goldbook.A00532
- ^ a b Stumm, Werner; Morgan, James (1996). Aquatic Chemistry. Chemical Equilibria and Rates in Natural Waters (3rd ed.). King John Wiley & Sons, Inc. ISBN9780471511847.
- ^ Harned, H. S.; Owen, B. B. (1958). The Physical Chemistry of Electrolytic Solutions (3rd ed.). New York: Reinhold. pp. 635.
- ^ Multinational Association for the Properties of Water and Steam (IAPWS)
- ^ Bandura, Andrei V.; Lvov, Serguei N. (2006). "The Ionisation Constant of Body of water over Wide Ranges of Temperature and Density" (PDF). Journal of Physical and Chemical Reference Data. 35 (1): 15–30. Bibcode:2006JPCRD..35...15B. doi:10.1063/1.1928231.
- ^ 0.1 MPa for T < 100 °C. Saturation insistence for T > 100 °C.
- ^ Harned, H. S.; Owen, B. B. (1958). The Physical Chemistry of Electrolytic Solutions (3rd ed.). New York: Reinhold. pp. 634–649, 752–754.
- ^ Lide, D. R., erectile dysfunction. (1990). CRC Handbook of Chemistry and Natural philosophy (70th male erecticle dysfunction.). Boca Raton (FL):CRC Press.
- ^ Geissler, P. L.; Dellago, C.; Chandler, D.; Hutter, J.; Parrinello, M. (2001). "Autoionization in liquid water". Science. 291 (5511): 2121–2124. Bibcode:2001Sci...291.2121G. CiteSeerX10.1.1.6.4964. doi:10.1126/science.1056991. PMID 11251111.
- ^ Manfred Eigen, M.; De Maeyer, L. (1955). "Untersuchungen über die Kinetik der Neutralisation I" [Investigations connected the kinetics of neutralization I]. Z. Elektrochem. 59: 986.
- ^ Stillinger, F. H. (1975). Theory and Building block Models for Water. Adv. Chem. Phys. Advances in Chemical Physics. 31. pp. 1–101. doi:10.1002/9780470143834.ch1. ISBN9780470143834.
- ^ Rapaport, D. C. (1983). "Hydrogen bonds in water". Mol. Phys. 50 (5): 1151–1162. Bibcode:1983MolPh..50.1151R. doi:10.1080/00268978300102931.
- ^ Chen, S.-H.; Teixeira, J. (1986). Social organization and Dynamics of Low-Temperature Piss as Premeditated by Scattering Techniques. Adv. Chem. Phys. Advances in Material Physical science. 64. pp. 1–45. doi:10.1002/9780470142882.ch1. ISBN9780470142882.
- ^ Tinoco, I.; Sauer, K.; Wang, J. C. (1995). Physical Chemistry: Principles and Applications in Natural Sciences (3rd ed.). Prentice-Residence hall. p. 386.
Extraneous links [edit out]
- General Chemical science – Autoionization of Water
what are the products of self ionization of water
Source: https://en.wikipedia.org/wiki/Self-ionization_of_water


![FIX: My printer does not print the whole page [HP, Canon]](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_vZFtj_c-ED5_8dwIv9sldr4UpNbYXNjXjtU40FTLMYwqSb-RLy6_YT1fopZ7TXssp_nqw_HheEr7UdQ7Ot3SjQI69wHtgIZD8X6KE04j3KrpXm1zfoGRB3T27dpop2lnezuVStHMW6NlMWx3s_GEvJAOw9Z4GILRHVlOdzvxuVkpBbrl6PQohM2f7g7g=w680)
![OneDrive files not syncing on iPad or iPhone? [Full Fix]](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_tYSgJ2OrRkNlFlTlNmAAtgrVjaWcPn0WIPWb2MDryPO2dtRdP2eBTCkqbXspWG8h1uNFOvBvQyH4MPHRUatkseFYkIIxzFCptz1k6rqrm54_8ATMDea_TTl6o9x__DdG9b51w6x7Ad5w=w680)

0 Komentar